
- Lunes, marzo 31, 2025
NIÑOS “PIEL MARIPOSA”
Academia Mexicana de Ciencias
Boletín AMC/438/14
México, D.F., 22 de diciembre de 2014
- Se trata de una enfermedad de la piel genética hereditaria sin cura hasta el momento, pero cada vez más se avanza en la investigación científica para conocer sus causas y eventual tratamiento
- En México se desconoce el porcentaje de niños que nacen con enfermedades genéticas ampollosas
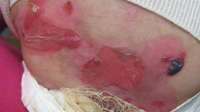
- Heridas y ampollas originadas por epidermólisis bullosa.
Foto: Cortesía del investigador, doctor Julio César Salas Alanís.
Imagen en alta resolución
Caracterizada por una marcada fragilidad de la piel y mucosas de todo el cuerpo provocando la formación de ampollas y úlceras de manera espontánea o bien ante el menor traumatismo, la epidermolisis bullosa (EB) es una enfermedad de la piel hereditaria que se presenta en el nacimiento y no tiene cura, por lo que científicos de la Facultad de Medicina de la Universidad Autónoma de Nuevo León profundizan en sus estudios sobre las mutaciones de los genes causantes de este padecimiento con el objetivo de mejorar la calidad de vida de los pacientes.
“Este proyecto comenzó en 1994 y desde entonces hemos tratado de detectar las mutaciones que afectan a la población mexicana”, comentó el doctor Julio César Salas Alanís, investigador líder del proyecto, quien agregó que estos trastornos congénitos están caracterizados por deficiencias o mal funcionamiento de las proteínas que mantienen unida la epidermis con la dermis o, incluso, a las uniones que se encuentran dentro de la epidermis.
La frecuencia de esta enfermedad a nivel nacional no se conoce, comentó el especialista en dermatología, sin embargo la Fundación DEBRA México -organización de la que es fundador y está dedicada a promover la investigación y la educación sobre la epidermolisis bullosa- registra alrededor de 500 familias con algún antecedente o integrante afectado con este padecimiento tan solo en la zona norte de México. Se estima que alrededor del mundo hay 500 mil enfermos.
El protocolo para el seguimiento de la enfermedad en las personas comienza a partir de la realización de la historia clínica detallada para orientar el diagnóstico inicial. “Posteriormente se practica una biopsia de piel, se tiñe con inmunoflorecencias cutánea directas para medir la cantidad de proteína faltante y/o defectuosa en la piel y con el objetivo de detectar las mutaciones se toma sangre periférica para extraer el ADN (ácido desoxirribonucleico) de los pacientes. Se buscan las mutaciones por medio de la secuenciación automática directa utilizando técnicas de PCR (reacción en cadena de la polimerasa) para amplificar el ADN y también se recurre a estudios de haplotipos”, explicó el también integrante de la Academia Mexicana de Ciencias y de la Academia Nacional de Medicina.
La EB es un padecimiento –conocido también como ¨piel mariposa o ¨niños de cristal”- en el que están involucrados 18 pares de genes. Cada uno de los genes dicta la producción de una proteína específica en la piel, pero cuando falta uno de los dos genes o los dos, la enfermedad se presenta. “Existen cuatro tipos principales de esta enfermedad: simple, de unión, distrofia y mixta. Cada tipo tiene una proteína faltante específica; por ejemplo, en la forma distrofia falta la proteína de colágeno VII, mientras que en la simple, la más benévola, hay alteraciones en las citoqueratina 5 y 14”. Es debido a esta especificidad que aún no existe un blanco terapéutico que sea eficiente, por lo que el único camino a seguir por ahora es un diagnóstico oportuno.
Con este objetivo, Salas Alanís ha podido detectar hasta ahora 36 familias mexicanas que reportan mutaciones en sus genes para el tipo distrofia. También informó que lleva a cabo un trabajo en coordinación con la Universidad de Jefferson, en Estados Unidos, para realizar una detección masiva de mutaciones en todas las familias mexicanas en nuestro país. El propósito del proyecto es acrecentar el banco de ADN que inició el especialista hace 20 años y el cual cuenta hasta ahora con alrededor de unas 400 muestras de genes de familias mexicanas.
Para identificar a la EB en etapas tempranas, el equipo de científicos colaboradores de Salas Alanís desarrolló un sistema de diagnóstico durante el embarazo. “Si hay antecedentes de un paciente en una determinada familia y conocemos el gen y su mutación causante, durante el siguiente embarazo podemos saber si el producto estará enfermo o no a través de una biopsia de la vellosidad coriónica –que presentan la misma constitución genética que el feto en desarrollo y pueden analizarse para detectar anomalías cromosómicas y genéticas-, se trata de una prueba que consiste en tomar una muestra de placenta para saber si el producto porta la enfermedad o no”.
De esta manera, se busca anticipar la aparición del padecimiento detectando a las familias o individuos portadoras de este gen para brindarles información y ayuda.
La Fundación DEBRA México AC (Dystrophic Epidermolysis Bullosa Research Association), realiza protocolos de investigación básica y clínica, y apoya a enfermos con epidermolisis bullosa congénita.
Entre las líneas de investigación del doctor Salas Alanís están también las de genética y psoroasis, hipetricosis universal congénita, enfermedades ampollosas autoinmunes y herpes genital.
Mariana Dolores.
Regresar Arriba, o a Comunicados, o al Inicio.
AMC "Casa Tlalpan" Calle Cipreses s/n, km 23.5 de la carretera federal México - Cuernavaca, San
Andrés Totoltepec, Tlalpan, C.P. 14400, México, D.F.
Coordinación de Comunicación y Divulgación
Teléfonos: (52-55) 58 49 49 04, Fax: (52-55) 58 49 51 10, amcpress@unam.mx
Mapa de ubicación
{kind=link}