
- Sábado, marzo 29, 2025
Científicos mexicanos monitorean comportamiento del nuevo coronavirus
Academia Mexicana de Ciencias
Boletín AMC/051/20
Ciudad de México, 24 de abril de 2020
- El proyecto a mediano plazo, seis meses, consiste en realizar la secuenciación del SARS-CoV-2 de al menos 250 personas que hayan presentado la enfermedad COVID-19.
- Fundamental mantener la vigilancia epidemiología y genómica del virus para identificar variantes que puedan circular de manera predominante en México al adaptarse a ciertas características ambientales, así como a determinantes genéticos y epidemiológicos de la población mexicana.
-
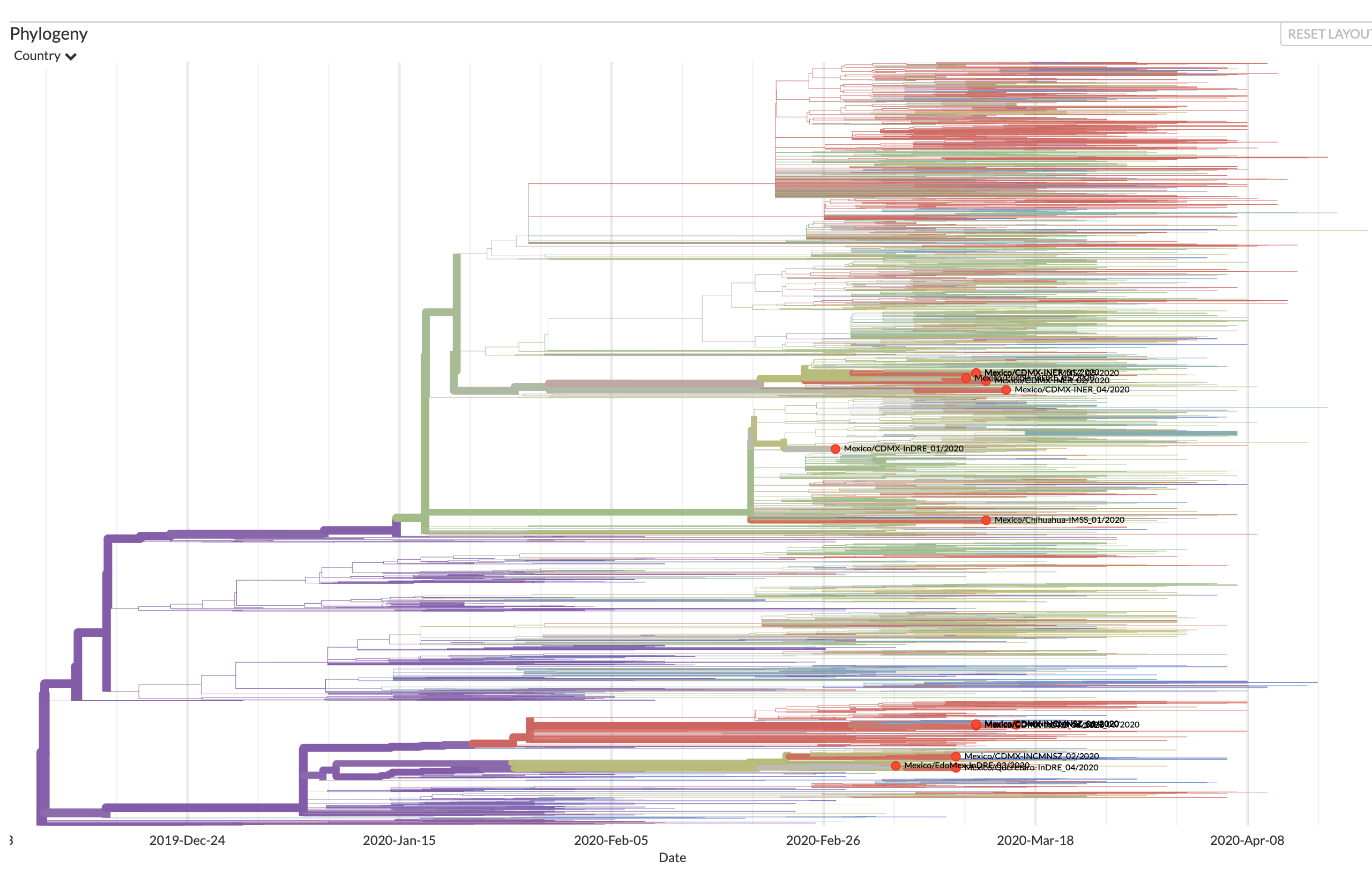
- Mapa filogenético de relaciones genéticas entre los genomas obtenidos de personas infectadas con SARS-CoV-2. Los puntos rojos muestran las secuencias mexicanas.
Imagen: Imagen tomada de Nexstrain.org.
Un grupo de investigadores mexicanos del Instituto de Diagnóstico y Referencia Epidemiológicos, el Instituto Nacional de Enfermedades Respiratorias, el Instituto Mexicano del Seguro Social, el Instituto Nacional de Ciencias Médicas y Nutrición y la Universidad Nacional Autónoma de México, así como de connacionales trabajando en la Universidad de Oxford, desarrolla un programa de secuenciación del genoma del SARS-CoV-2, con el apoyo del Consejo Nacional de Ciencia y Tecnología (Conacyt).
Los hallazgos
La primera secuencia del genoma del virus SARS-CoV-2 se obtuvo a partir de una muestra respiratoria del primer caso que se registró en el país. Después se secuenciaron otras 32 muestras con un enfoque metagenómico, pudiéndose obtener la secuencia del genoma completo del virus en 16 de ellas. De los 17 virus en total, 15 fueron de casos importación, uno de un apersona que tuvo contacto en el país con un viajero y uno más que puede ser atribuido al llamado contagio comunitario.
En un primer análisis, los investigadores encontraron que uno de los casos presentó un virus cuyo genoma fue esencialmente idéntico (sólo dos cambios) a otro caracterizado a partir de un caso importado, sin embargo, la primera persona no tuvo nada que ver con la persona que viajó, “lo que indica que el contagio comunitario pudo haber empezado a mediados del mes de marzo”, dijo Carlos Arias Ortiz, investigador adscrito al Departamento de Genética del Desarrollo y Fisiología Molecular del Instituto de Biotecnología de la UNAM.
Las secuencias del genoma de los 17 virus caracterizados muestran una mayor cercanía genética con los grupos del virus que estaba circulando en esas fechas en Europa, más que con los linajes virales que surgieron más tempranamente en China y que se distribuyeron rápidamente en el sureste asiático. Hasta ahora, se han identificado tres grupos o linajes del SARS-CoV-2 en el mundo, pero las muestras que analizaron en México están distribuidas casi mitad y mitad en dos de ellas, la G y S, no se han encontrado del grupo V.
“Toda esta investigación sirve para tener una base de conocimiento de la secuencia del genoma de algunos de los primeros virus que entraron al país, de una parte de ellos no tenemos todo el registro, pero cuando menos sabemos que este subconjunto de virus llegó a México entre el 10 y 15 de marzo”, señaló el también integrante de la Academia Mexicana de Ciencias.
En breve se realizará la secuenciación de 60 muestras más, los investigadores continuarán con el análisis de datos hasta llegar a la secuenciación de 250 muestras, de las cuales se espera obtener al menos 150 genomas completos. “Tenemos recursos para las 250 secuenciaciones, otorgados por el Conacyt, pero esperamos poder ampliar este proyecto un par de años para vigilar a más largo plazo la evolución del virus en el país, queremos seguir la pista de cómo se comporta en la población mexicana, cuáles son los variantes del virus que pudieran seleccionarse y adquirir cambios que pudieran impactar sus características biológicas, y para esto necesitaremos más recursos”, dijo el experto en virología.
¿Qué se puede rastrear?
El equipo de investigadores se encargará de observar y analizar el comportamiento del virus a lo largo del tiempo, los primeros resultados de las muestras secuenciadas con éxito, aunque es pequeña, no es despreciable. Estos resultados se compararán con los hallazgos del siguiente grupo de muestras de las que se logre obtener el genoma completo y así sucesivamente.
“Es como armar una película con fotogramas, este primer resultado apenas es una pieza de esta película continua que empezó en Wuhan, China. Conforme avance la investigación en nuestro país y en el mundo, tendremos mayor conocimiento”.
El también integrante de la Sociedad Mexicana de Virología explicó que esta vigilancia es muy necesaria porque a lo largo del tiempo los virus de RNA, de una sola hebra, como el coronavirus, van modificando su genoma y lo que están haciendo es monitorear esos cambios, porque les podrá decir si uno de los primeros 17 virus cuyos genomas se secuenciaron originalmente, o alguno de los que se secuenciarán posteriormente, empieza a ser predominante en la población, lo que pudiera estar relacionada con factores como el medio ambiente, la genética mexicana, o a la adaptación para replicarse en humanos de manera general, entre otros.
El método para obtener el genoma tarda tres días, desde que se tiene la muestra lista en el laboratorio para realizar el procedimiento y hasta que se tiene el análisis de los datos de la secuenciación. Cada secuenciación tiene un costo de aproximadamente cinco mil pesos.
Explicó que el genoma del SARS-CoV-2 tiene aproximadamente 30 mil nucleótidos, pequeños ladrillos o bloques que conforman su estructura, los virus que secuenciamos presentan entre 8 y 14 cambios de nucleótidos en relación al primer virus que se secuenció en Wuhan, por eso se dice que los virus que se presentaron en el país muestran una alta conservación, un mínimo de identidad de 99.97% con la cepa original.
La filogenómica del SARS-CoV-2, que puede consultarse en nextstrain.org, una página que compila el análisis de alrededor de 3,500 genomas de más de 40 países, informa que dependiendo de dónde los virus presenten los cambios (cada muestra de virus presenta cambios en diferentes partes del genoma) se clasifican en linajes, y este árbol familiar del coronavirus tiene tres grandes ramas, que son los grupos antes mencionados (G, S y V). “La buena noticia es que, a pesar de ser el virus de RNA cuyo genoma es el más grande que se conoce, tiene una maquinaria molecular que le permite corregir los errores que introduce la RNA polimerasa viral durante la replicación del genoma, lo que se refleja en el hecho de que los cambios reportados oscilan sólo entre 8 y 14, es decir, es un virus más estable que otros virus de RNA, como el virus de la influenza”.
“El equipo de científicos, que está integrado por alrededor de 18 personas, desarrolla el proyecto Epidemiología Genómica del SARS-CoV-2 en México, y aunque el alcance con 250 muestras, es limitado, es importante”, reconoció Arias.
Añadió que es muy importante continuar vigilando la evolución del genoma del virus para detectar mutaciones que pudieran asociarse a cambios en su comportamiento biológico (niveles de contagio, virulencia, estabilidad), a la aparición de variantes resistentes a fármacos, y variantes que disminuyan la eficacia de las vacunas, una vez que estas medidas de prevención y tratamiento hayan sido aprobadas y se apliquen.
Elizabeth Ruiz Jaimes
Regresar Arriba, o a Comunicados, o al Inicio.
AMC "Casa Tlalpan" Calle Cipreses s/n, km 23.5 de la carretera federal México - Cuernavaca, San
Andrés Totoltepec, Tlalpan, C.P. 14400, México, D.F.
Coordinación de Comunicación y Divulgación
Teléfonos: (52-55) 58 49 49 04, Fax: (52-55) 58 49 51 10, amcpress@unam.mx
Mapa de ubicación
{kind=link}